
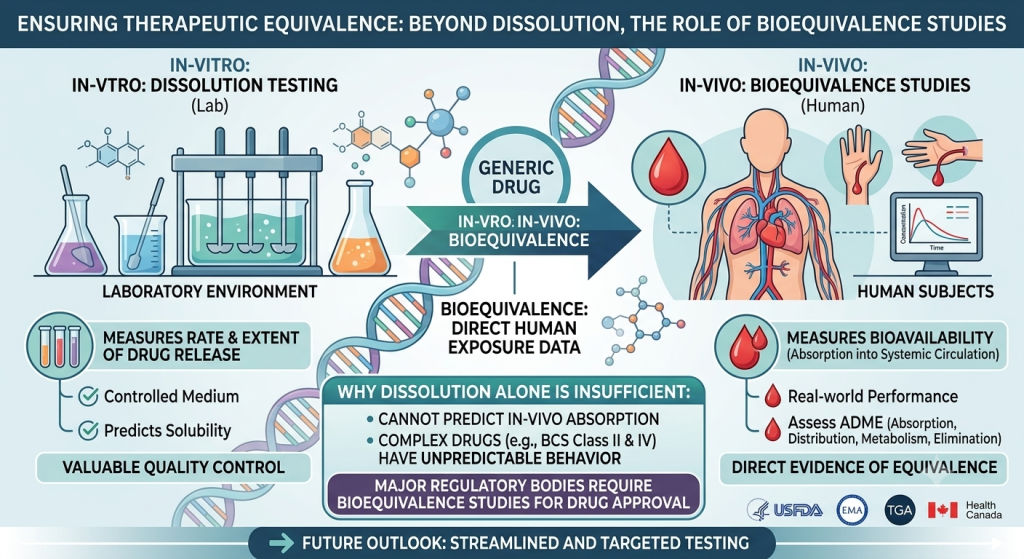
Generic medicines play a vital role in improving access to affordable healthcare worldwide. Compared with innovator or reference products, generic drugs are generally available at a lower cost, making them an important option for patients and healthcare systems. However, regulatory authorities must ensure that generic medicines meet the same standards of quality, safety, and efficacy as the reference products. In highly regulated markets such as the U.S. Food and Drug Administration (USFDA), European Medicines Agency (EMA), Therapeutic Goods Administration (TGA), and Health Canada, bioequivalence studies are generally required for generic drug approval. In contrast, some low- and middle-income countries have historically granted approval for certain generic products based primarily on in-vitro dissolution data because of socioeconomic considerations, limited resources, and the high cost associated with conducting bioequivalence studies.
A bioequivalence study is a pharmaceutical investigation designed to compare the bioavailability of a generic product with that of a reference product in human subjects. The study evaluates whether both products deliver the active ingredient into the systemic circulation at a similar rate and extent. Demonstrating bioequivalence provides evidence that the generic medicine is expected to produce the same therapeutic effect and safety profile as the reference product. Dissolution testing, on the other hand, is an in-vitro quality control test that measures the rate and extent to which a drug substance dissolves in a specified medium under controlled laboratory conditions. Dissolution is an important step because a drug generally must dissolve before it can be absorbed into the body. Therefore, dissolution testing is widely used during product development, manufacturing, and quality assessment.
Although dissolution testing is valuable, it cannot fully predict how a medicine will behave in the human body. A dissolution test only evaluates drug release under laboratory conditions and does not directly measure drug absorption, distribution, metabolism, or elimination. Consequently, similar dissolution profiles do not always guarantee similar in-vivo performance. For these reasons, major regulatory agencies generally require bioequivalence studies because they directly assess drug exposure in humans. Bioequivalence studies provide evidence that the generic product delivers the active ingredient to the bloodstream at a rate and extent comparable to the reference product, thereby supporting therapeutic equivalence. Nevertheless, bioequivalence studies are expensive, time-consuming, and require significant human and financial resources. These challenges can be particularly burdensome for manufacturers and regulatory agencies in low-income countries. As a result, reliance on dissolution testing may help reduce development costs and improve the availability of affordable medicines. However, this approach may not be appropriate for all drug products. Particular attention should be given to drugs belonging to the Biopharmaceutical Classification System (BCS) Class II and Class IV categories, where drug absorption is often influenced by poor solubility and other complex factors. For such products, dissolution testing alone may not provide sufficient assurance of therapeutic equivalence. In these cases, bioequivalence studies should be strongly considered to ensure patient safety and treatment effectiveness.
Future Perspective:
As pharmaceutical science and regulatory systems continue to advance, low- and middle-income countries may benefit from adopting a more risk-based approach to generic drug approval. While dissolution testing can remain an important and cost-effective tool, greater emphasis should be placed on conducting bioequivalence studies for products with a higher risk of variable absorption or therapeutic failure. Strengthening regional bioequivalence study centers, improving regulatory capacity, and encouraging international collaboration could help reduce study costs and increase accessibility. In the future, a balanced approach that combines affordability with scientifically robust evidence will be essential to ensure both access to medicines and public health protection.
Author:
Roshan Gyawali /Member of Dirgha Research Group
Assistant Manager,
Biogain Remedies Pvt. Ltd., Rupandehi, Nepal
Email: [email protected]



